在创新药物研发中,一个核心问题是如何快速发现具有理想生物学特性的新化学实体。然而,满足Lipinski's类药性的化学空间所包含的理论分子数可能超过1063【1】,如何更高效地探索这样巨大的多样性空间,找到具有良好的可合成性和成药性的新分子是摆在计算化学家和药物化学家面前的一道难题。针对这一问题,Reymond团队利用穷举法构建重原子数在13到17范围之内的分子结构,建立了包含有166亿个化合物的GDB库【2】;Levré等人从市售的炔烃和叠氮化物出发通过使用Click反应规则构建了Zinclick数据库【3】,该库具有较好的可合成性但结构类型相对单一;此外,也有一些基于人工智能(Artificial Intelligence,AI) 方法,例如Segler等人利用LSTM模型设计集中化合物库【4】,Zhavoronkov等人将基于生成对抗网络 (GANs)的AI技术应用到化学空间探索【5】。虽然方法众多,但这些虚拟化合物库都面临着多样性、可合成性和成药性的多方面问题。如果将寻找药物新化学实体看做数据科学中的采样问题,那在已知药物结构所代表的数据点附近进行采样无疑是获得类药分子结构的高效策略之一。
近日,药物科学与技术学院(以下简称药学院)蒋华良院长、郑明月研究员工作室从已批准上市药物出发,采用电子等排体和化学反应转化规则构建了成药性拓展空间数据库DrugSpaceX (https://drugspacex.simm.ac.cn/)。研究结果近期在线发表于Nucleic Acids Research,题为“DrugSpaceX: a large screenable and synthetically tractable database extending drug space” 。

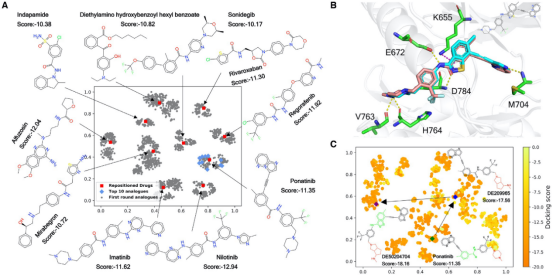
目前版本的DrugSpaceX包含超过1亿种可用于虚拟筛选的新分子结构,且在类药性、可合成性和三维化学多样性空间覆盖率方面均具有突出的特点,为开展虚拟筛选和药物分子设计提供了高质量的资源。此外,DrugSpaceX还提供了几个规模较小的子集,包括10%多样性子集,扩展的类药性子集,类药性子集,先导化合物子集和片段子集等,可供用户免费下载使用。研究团队使用了盘状蛋白结构域受体1 (discoidin domain receptor 1, DDR1) 进行案例研究,展示了如何利用DrugSpaceX快速筛选活性化合物 (图2)。首先,以药物数据集为出发点进行基于结构的虚拟筛选,选择分子对接打分前十的药物分子。通过文献检索可以发现其中Imatinib,Nilotinib,Ponatinib均对DDR1有交叉活性。然后,从DrugSpaceX上检索前十名药物分子第一轮衍生物,再进行第二轮筛选。可以发现,在第一轮衍生物中对接打分前十的化合物主要集中在ponatinib周围(如图2A所示),其中排名第3的化合物DE209841,已被Insilico Medicine最近报道的DDR1抑制剂专利所覆盖(NO. WO2020079652A1)。图2B中显示了DE209841的预测结合模式,与Zhavoronkov等人文章报道的结合模式吻合【5】。进一步解析重构数据集并采用相同筛选流程,可以发现对接打分和配体效率更高的新结构DE50204704。如图2C所示,该分子结构可以视作是老药Ponatinib经过对“tail” 和“linker” 片段进行两轮改造得到的。
综上,DrugSpacesX除了帮助药物化学家能够进行快速的骨架跃迁和分子设计, 为我们提供了一种高效探索类药化学空间的思路。可以发现,通过将专家知识和人工智能相互融合,我们可以在巨大的虚拟化学空间中更容易地找到具有理想生物效应的目标化合物。此外,Christoph Gorgulla等人近期在Nature发表的文章中也指出超大规模虚筛可以提高真阳性率【7】,DrugSpaceX也可以与VirtualFlow等虚拟筛选平台结合使用,通过扩大初始筛选规模和提高筛选库质量两方面来进一步提升效率。目前,研发团队还在对DrugSpaceX进行扩充和完善,期待后续可以推出功能更为强大的版本。
药学院博士生杨天标和上海药物所博士后李召军为共同第一作者,药学院蒋华良院长及其工作室郑明月研究员为本论文共同通讯作者。该工作得到国家新药重大专项和科学院先导专项的资助。
原文链接:https://doi.org/10.1093/nar/gkaa920
参考文献:
1.Reymond, J.L. (2015) The chemical space project. Acc. Chem. Res., 48, 722-730.
2.Ruddigkeit, L., van Deursen, R., Blum, L.C. and Reymond, J.L. (2012) Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model., 52, 2864-2875.
3.Levré, D., Arcisto, C., Mercalli, V. and Massarotti, A. (2019) ZINClick v.18: Expanding Chemical Space of 1,2,3-Triazoles. J. Chem. Inf. Model., 59, 1697-1702.
4.Segler, M.H.S., Kogej, T., Tyrchan, C. and Waller, M.P. (2018) Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent Sci, 4, 120-131.
5.Zhavoronkov, A., Ivanenkov, Y.A., Aliper, A., Veselov, M.S., Aladinskiy, V.A., Aladinskaya, A.V., Terentiev, V.A., Polykovskiy, D.A., Kuznetsov, M.D., Asadulaev, A. et al. (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol., 37, 1038-1040.
6.Yang, T., Li, Z., Chen, Y., Feng, D., Wang, G., Fu, Z., Ding, X., Tan, X., Zhao, J., Luo, X. et al. (2020) DrugSpaceX: a large screenable and synthetically tractable database extending drug space. Nucleic Acids Research.
7.Gorgulla, C., Boeszoermenyi, A., Wang, Z.F., Fischer, P.D., Coote, P.W., Padmanabha Das, K.M., Malets, Y.S., Radchenko, D.S., Moroz, Y.S., Scott, D.A. et al. (2020) An open-source drug discovery platform enables ultra-large virtual screens. Nature, 580, 663-668.
 国科大杭高院药学院
国科大杭高院药学院